Abstract
FABRY DISEASE AND TREATMENT-AN OVERVIEW
T. Rajeshwari*, V. Manjusha
Fabry disease (FD) is an X-linked, hereditary, lysosomal storage disease caused by deficiency of enzyme α- galactosidase A (α-Gal A), which results in accumulation of neutral glycosphingolipid globotriaosylceramide(Gb3) in walls of small blood vessels, nerves,dorsal root ganglia, renal glomerular and tubular epithelial cells and cardiomyocytes. Males are mostly affected whereas women mainly act as carriers. Onset of FD symptoms depends upon its clinical type which vary from neuropathic pain, hypohidrosis, gastrointestinal symptoms, and angiokeratomas, chronic kidney disease, cardiomyopathy and cerebral events as seen in adults to severe mental retardation as seen in infantile form which drastically affects the quality of life and reduces life span of affected individuals. Enzyme replacement therapy (ERT) with intravenous infusions of recombinant human α-galactosidase A is approved by FDA for treating FD. Alternative treatments under investigation include substrate reduction therapy (SRT), chaperons, stem cell transplant and gene therapy.The aim of present study is to review pathophysiology, clinical manifestations, diagnosis, treatment of FD.
Manuscript Submission
Submit your manuscript at Online Submission System
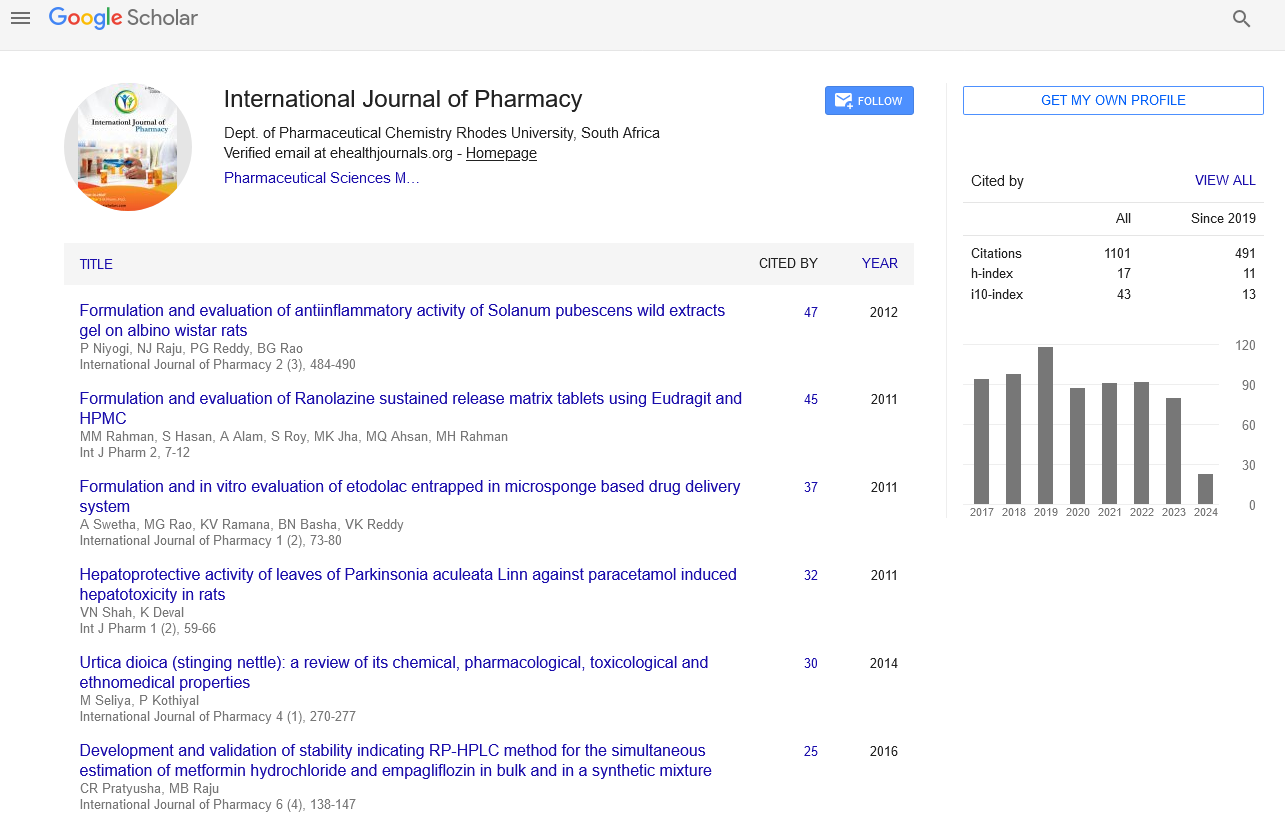
Google scholar citation report
Citations : 1101
International Journal of Pharmacy received 1101 citations as per google scholar report
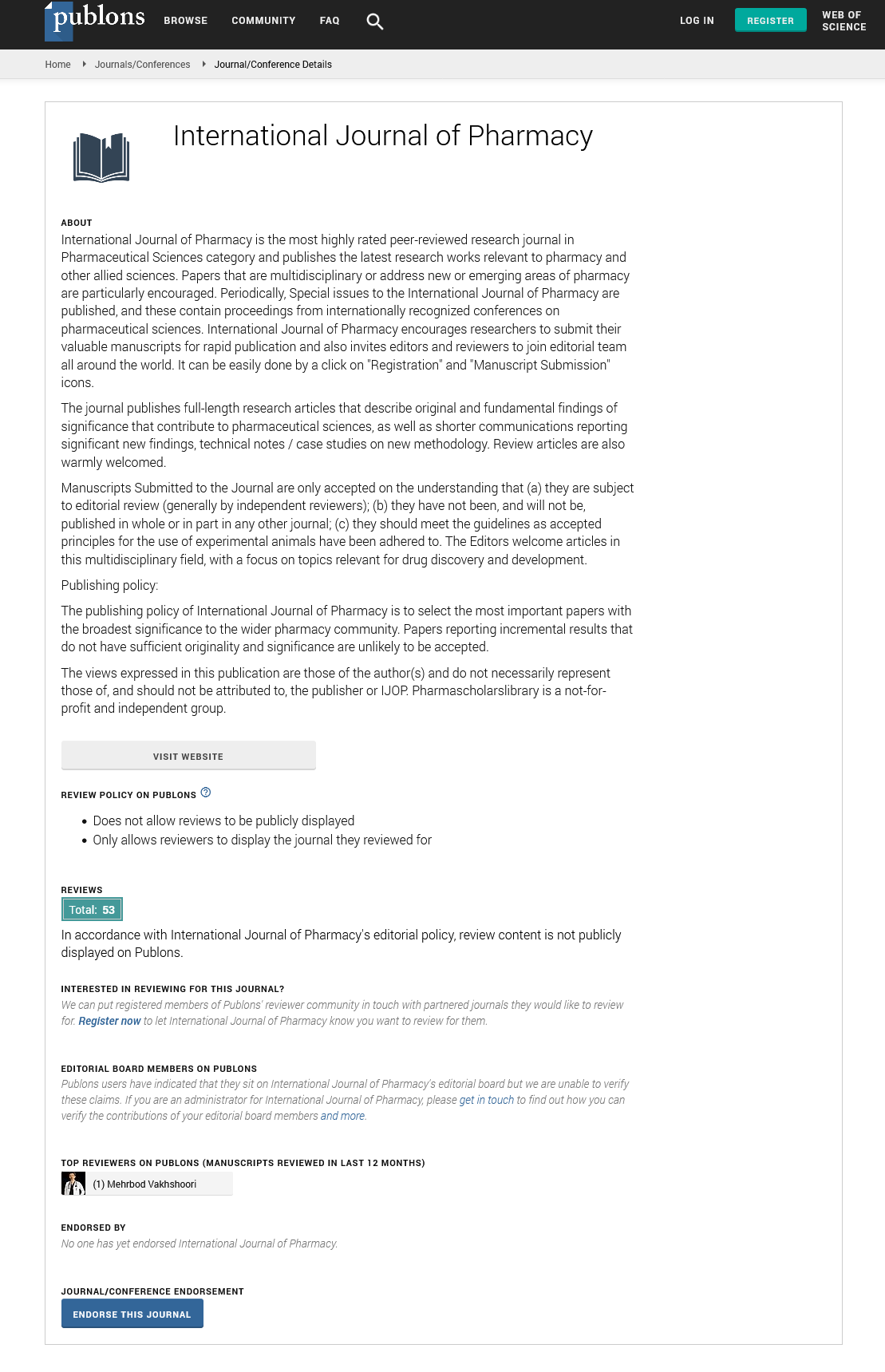
International Journal of Pharmacy peer review process verified at publons
Indexed In
- CAS Source Index (CASSI)
- HINARI
- Index Copernicus
- Google Scholar
- The Global Impact Factor (GIF)
- Polish Scholarly Bibliography (PBN)
- Cosmos IF
- Open Academic Journals Index (OAJI)
- Directory of Research Journal Indexing (DRJI)
- EBSCO A-Z
- OCLC- WorldCat
- MIAR
- International committee of medical journals editors (ICMJE)
- Scientific Indexing Services (SIS)
- Scientific Journal Impact Factor (SJIF)
- Euro Pub
- Eurasian Scientific Journal Index
- Root indexing
- International Institute of Organized Research
- InfoBase Index
- International Innovative Journal Impact Factor
- J-Gate