HTML
Short Communication - (2022) Volume 12, Issue 3
Aspirin: An Effective Antiplatelet Drug
Cyrus Obama**Correspondence: Cyrus Obama, Department of Medicinal Chemistry, University of Uppsala, Uppsala, Sweden, Email:
Received: 08-Mar-2022, Manuscript No. IJP-22-61749; Editor assigned: 11-Mar-2022, Pre QC No. IJP-22-61749 (PQ); Reviewed: 29-Mar-2022, QC No. IJP-22-61749; Revised: 05-Apr-2022, Manuscript No. IJP-22-61749 (R); Published: 12-Apr-2022, DOI: 10.37532/2249-1848-2022.12(3).13
About the Study
Aspirin is a powerful antiplatelet drug. It inhibits cyclooxygenase (COX)-1 in platelets in an irreversible manner, reducing the synthesis of prostaglandin endoperoxides and thromboxane A2, a platelet agonist. In individuals with a history of cardiovascular, cerebrovascular, or peripheral vascular illness, aspirin has been proven to reduce the risk of subsequent thromboembolic events [1]. The antiplatelet medication aspirin has been examined the most. Aspirin decreases vascular death by 15% and nonfatal vascular events by 30% in high-risk individuals, according to more than 100 randomised trials.
Mode of action
The ability of aspirin to permanently block the COX activity of prostaglandin H-synthase-1 and prostaglandin H-synthase-2 is the bestunderstood mechanism of action (also referred to as COX-1 and COX- 2, respectively). The conversion of arachidonic acid to prostaglandin H2 is catalysed by COX isozymes, which is the first committed step in prostanoid production (PGH2). Thrombboxane A2 (TXA2) and PGI2 are both direct precursors of PGH2.
The acetylation of a strategically positioned serine residue (Ser529 in COX-1, Ser516 in COX-2) blocks the COX channel, blocking substrate access to the enzyme's catalytic site [2]. This is the molecular basis of aspirin's permanent suppression of COX activity.
Low dosages of aspirin (75-150 mg) given once daily can completely or nearly completely block platelet COX-1. Because nucleated cells rapidly resynthesize the enzyme, suppression of COX-2-dependent pathophysiologic processes (e.g., hyperalgesia, inflammation) necessitates higher dosages of aspirin and a much shorter dosing interval. When aspirin is used as an anti-inflammatory rather than an antiplatelet agent, 10 to 100 fold larger daily doses of the drug are necessary. Because aspirin's GI toxicity is dose dependent, its benefit/ risk profile is dose dependant..
PGH2 is processed by human platelets and vascular endothelial cells to create TXA2 and PGI2, respectively [3]. Platelet aggregation and vasoconstriction are induced by TXA2, whereas platelet aggregation and vasodilation are induced by PGI2. TXA2 is very susceptible to aspirin suppression because it is predominantly generated from COX-1 (mostly from platelets). Vascular PGI2, on the other hand, can be produced by COX-1, but COX-2 is the main source, even under physiological conditions. COX-1-dependent PGI2 synthesis is transitory and sensitive to aspirin suppression in response to agonist stimulation (e.g., bradykinin). COX-2-mediated PGI2 production occurs over time in response to laminar shear stress and is relatively unaffected by low-dose aspirin, which could explain why, despite transient suppression of COX-1-dependent PGI2 release, daily dosesof aspirin in the range of 30 to 100 mg still result in significant residual COX-2-dependent PGI2 biosynthesis. It is unknown whether the greater inhibition of PGI2 generation caused by larger aspirin doses is enough to cause or predispose to thrombosis. PGI2 appears to be thromboprotective, according to two lines of data. For starters, animals lacking the PGI2 receptor are more prone to injury-induced thrombosis. Second, the cardiovascular toxicity of COX-2 inhibitors supports the idea that PGI2 is crucial for thromboresistance when platelet TXA2 production is not adequately inhibited.
Pharmacokinetics of aspirin
In the stomach and upper intestine, aspirin is rapidly absorbed. Aspirin levels peak 30 to 40 minutes after intake, and platelet activity is inhibited within 1 hour. Enteric-coated aspirin, on the other hand, can take 3 to 4 hours to reach peak plasma levels following administration. If a quick effect is needed and only enteric-coated tablets are available, they should be chewed rather than eaten whole. Regular aspirin pills have a 40 percent to 50 percent oral bioavailability over a wide dosage range [4]. Enteric-coated tablets and sustained-release, microencapsulated formulations have been found to have much decreased bioavailability. When enteric-coated medicines are administered at modest doses, their decreased bioavailability and poor absorption from the higher pH environment of the small intestine may result in inadequate platelet inhibition, especially in heavier people. In order to accomplish selective reduction of platelet TXA2 production without decreasing systemic PGI2 synthesis, a controlled-release formulation and a transdermal patch with low systemic bioavailability have been created. The controlled-release formulation was used successfully in the Thrombosis Prevention Trial; however it is unclear whether it provides any advantages over regular aspirin.
Conclusion
Antiplatelet therapy can help prevent platelet-rich arterial thrombi from forming in high-shear situations. Antiplatelet drugs are less effective than anticoagulants for the prevention of fibrin-rich thrombi that form under low-shear conditions, such as VTE and left atrial appendage thrombi in patients with atrial fibrillation, but antiplatelet drugs are less effective than anticoagulants for these indications. The ability of aspirin to disrupt well-characterized platelet activation and aggregation pathways explains their thrombosis-prevention efficacy. These effects also cause bleeding, which is a common side effect of antiplatelet medication.
References
- Jacobsen AP, Raber I, McCarthy CP, et al. Circulation. 2020;142(16):1579-1590.
- Loll PJ, Picot D, Garavito RM. Nat Struct Biol. 1995;2(8):637-643.
- Sonnweber T, Pizzini A, Nairz M, Weiss G. Int J Mol Sci. 2018;19(11):3285.
- Patrono C, Ciabarroni G, Patrignani P, et al. Platelets Vasc Occ. 1985;72(6):1177-1184.
Manuscript Submission
Submit your manuscript at Online Submission System
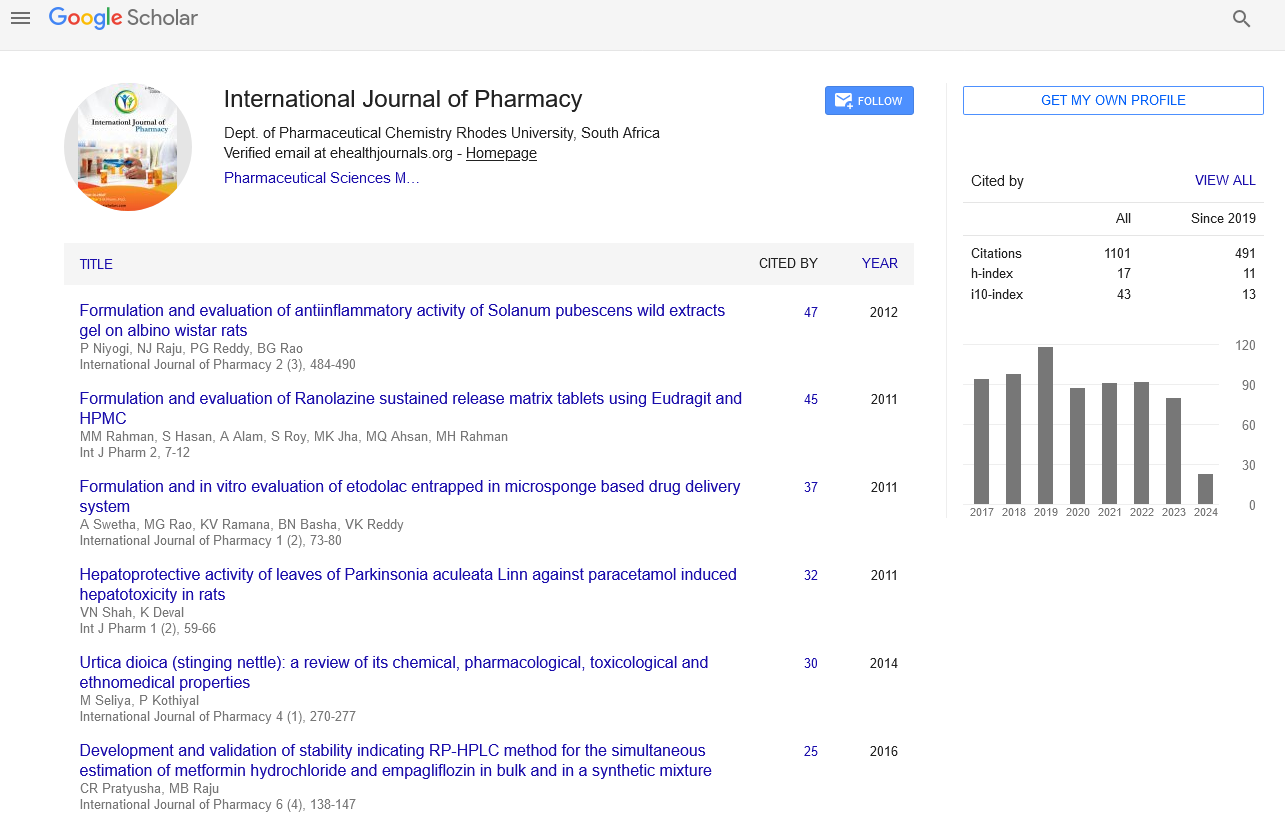
Google scholar citation report
Citations : 1101
International Journal of Pharmacy received 1101 citations as per google scholar report
International Journal of Pharmacy peer review process verified at publons
Indexed In
- CAS Source Index (CASSI)
- HINARI
- Index Copernicus
- Google Scholar
- The Global Impact Factor (GIF)
- Polish Scholarly Bibliography (PBN)
- Cosmos IF
- Open Academic Journals Index (OAJI)
- Directory of Research Journal Indexing (DRJI)
- EBSCO A-Z
- OCLC- WorldCat
- MIAR
- International committee of medical journals editors (ICMJE)
- Scientific Indexing Services (SIS)
- Scientific Journal Impact Factor (SJIF)
- Euro Pub
- Eurasian Scientific Journal Index
- Root indexing
- International Institute of Organized Research
- InfoBase Index
- International Innovative Journal Impact Factor
- J-Gate